Mission
Our mission is to support the University’s expectations of high quality research via quality assurance (QA) and quality improvement (QI) activities.
HRQA monitors and reviews active research protocols to ensure that the participants’ rights, safety and welfare are protected and that the research is conducted in accordance with the approved research plan and complies with all applicable Federal regulations and University policies.
The QA review process is an essential element of our accreditation by the Association for the Accreditation of Human Research Protection Programs (AAHRPP). WashU and AAHRPP recognize that the commitment to human research participation does not end with the approval of the project by the Institutional Review Board (IRB).
As an outgrowth of the monitoring activities, the HRQA program provides Quality Improvement support via tailored education, guidance and resources that will assist in the performance of high quality research.

- Phone: 314-747-5525
- Email: HRQA@wustl.edu
Jeneane Braden
Associate Vice Chancellor for Research Integrity and Ethics
- Phone: 314-747-4152
- Email: bradenj@wustl.edu
- Offices: Human Research Quality Assurance Program (HRQA)
Institutional Animal Care and Use Committee (IACUC)
Office of Research Integrity & Ethics (ORIE)
Export Control
Conflicts of Interest (COI)
Jessica Schnable
Manager, Human Research Quality Assurance Program
- Phone: 314-747-5525
- Email: j.schnable@wustl.edu
- Offices: Human Research Quality Assurance Program (HRQA)
Office of Research Integrity & Ethics (ORIE)
Associated Topic: Human Subjects Research
Contacts
Jessica Schnable
Manager, Human Research Quality Assurance Program
j.schnable@wustl.edu
314-747-5525
Ian Brown
Research Compliance Specialist
ibrown@wustl.edu
314-747-6588
Megan Dandurand
Human Subjects Research QA/QI Program Analyst
dandurand@wustl.edu
314-747-5526
HRQA On-Demand
HRQA provides educational presentations and services tailored to the needs of your department, division, or research group. See the below menu for a list of presentation topics and services.
We can arrange presentations or group sessions on the following topics or any topic relevant to human subjects research.
You can request a presentation by clicking “Request Presentation” below the topic of interest in the following menu. If your preferred topic is not listed below please contact the program at HRQA@wustl.edu to request a topic specific presentation.
HRQA Education/Services
Services
- Consultations on best-practices, advice, or guidance on human research related matters
- Source document development/documentation guidance & best practices
- FDA Audit Prep Assistance
- PI Requested monitoring reviews
Education
- Good Clinical Practice Training
- Study Initiation Program Visit
- Case Studies in Compliance
- FDA audit readiness & lessons learned
- How to conduct internal audits – interactive activity
- Best Practices for Conducting Human Subjects Research
- Compliance trends
- Introduction to Human Subjects Research
- Topic specific education by request
FAQs
What research protocols are subject to review by the program?
HRQA Review Objectives
In order to affectively assess WashU’s entire human subjects portfolio, the HRQA program strives to complete at least one monitoring review within each school, every fiscal year as possible. Schools/departments with higher volumes of human subjects research may be reviewed more frequently. Studies are selected for review from each risk group to represent the different types of research.
Typical Studies that fall within HRQA Review
- Research studies with little to no outside oversight such as investigator-initiated, grant funded or non-funded human research studies.
- Then studies selected for review are categorized into 4 groups using a risk stratification model.
- Individual study components are assessed (based on how they are applied in the research) for potential risks to participants, PI/University and then classified into a risk group.
- Typically, most research components fall into more than one risk group.
- Studies with a greater and more cumulative potential risk are prioritized for review.
- Reviews may also be University directed or PI requested, regardless of risk group.
- Then studies selected for review are categorized into 4 groups using a risk stratification model.
- Typically the following types of study are excluded from selection unless group IV risk factors identified:
- Exempt Research
- Studies under the purview of Siteman Cancer Center (SCC) QASMC review.
- Studies with outside monitoring (e.g. clinical studies sponsored by industry with on-site and/or remote monitoring plan)
- PI’s conducting their first IND/IDE research project and/or with little experience conducting IND/IDE research, regardless of sponsorship or outside monitoring, may be subject to review due to high probability of FDA audit.
- The program will randomly select investigators that fall within this category throughout the fiscal year (5 to 10%) pending available projects.
- PI’s conducting their first IND/IDE research project and/or with little experience conducting IND/IDE research, regardless of sponsorship or outside monitoring, may be subject to review due to high probability of FDA audit.
Risk model related to study selection
Studies selected for potential monitoring are identified by reviewing the individual study components and evaluating how that particular study falls within the risk stratification model.
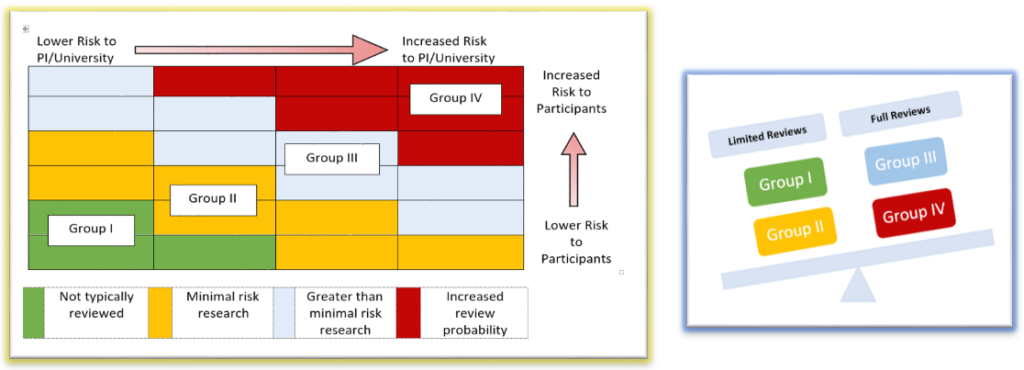
Types of Review Processes
Criteria for limited or full review are applied based on the risk assessment.
- Full Reviews are a comprehensive review of all research records related to the study to assess whether compliance standards have been met. Typically, full reviews are greater than minimal risk and have study components that primarily fall into risk Groups III or IV
- Assess whether compliance standards have been met in the following categories:
- Study enrollment status
- Execution of informed consent/assent
- Maintenance of privacy & confidentiality
- Participant eligibility
- Documentation of interventions/observations and study procedures
- Safety monitoring
- Regulatory documentation/multisite management
- Required education and HIPAA training
- Assess whether compliance standards have been met in the following categories:
- Limited Reviews are typically minimal risk and have study components that primarily fall into risk Groups I or II:
- When the primary risk is related to the execution of informed consent/assent (e.g. registries, repositories, observational, teaching studies, complicated consenting processes).
- Investigator initiated, rapidly enrolling and high enrolling studies
- Studies enrolling vulnerable populations (e.g. due to additional regulatory requirements, prisoners, wards of the state, participants not cognitively able to provide consent for themselves)
- Assess whether compliance standards have been met; primarily focuses on monitoring:
- Study enrolment status
- Execution of informed consent/assent; and
- Required education and training.
- When the primary risk is related to the execution of informed consent/assent (e.g. registries, repositories, observational, teaching studies, complicated consenting processes).
Risk Stratification Model
How will I be notified that my research is selected for monitoring?
The Principal Investigator will be notified via email that a particular study has been selected for review. The HRQA staff will then make arrangements to schedule a mutually agreeable appointment for on-site review, typically within 7-10 days of notification.
If the investigator does not comply with the request to schedule the audit, the Office of Research Integrity and Ethics/Vice Chancellor for Research Office will be notified of problems in scheduling the audit in a timely manner. If warranted, for-cause audits may be held without prior notice.
Does the principal investigator need to be present for the entire process?
After a short general discussion of the research protocol and how the research files are organized, the HRQA staff will need a place to work and access to the requested files. The Principal Investigator (PI) will need to meet with the HRQA staff at some point during or after the review process. It is not necessary for the PI or a research team member to be available throughout the entire review. Generally, someone who is able to answer questions should be available via phone or email.
What will be reviewed?
Depending on the type of review (limited or full) the review will assess whether compliance standards have been met in the following 8 categories: 1) Study Enrollment Status; 2) Execution of Informed Consent/Assent; 3) Maintenance of Privacy & Confidentiality; 4) Participant Eligibility; 5) Documentation of Interventions/Observations and Study Procedures; 6) Safety Monitoring; 7) Regulatory Documentation/Multisite Management; 8) Required Education and HIPAA Training.
The HRQA staff will need access to ALL participants’ electronic and hardcopy research records, regulatory documentation, and if applicable, specimens and specimen collection records. A sampling of the participants’ records will be reviewed based on the accrual to date and will span the course of enrollment.
The HRQA staff may contact subjects who have participated in the study in order to review their understanding of the consent process, the research project and their impressions of participation in the research process.
Who will receive the results of the monitoring activity?
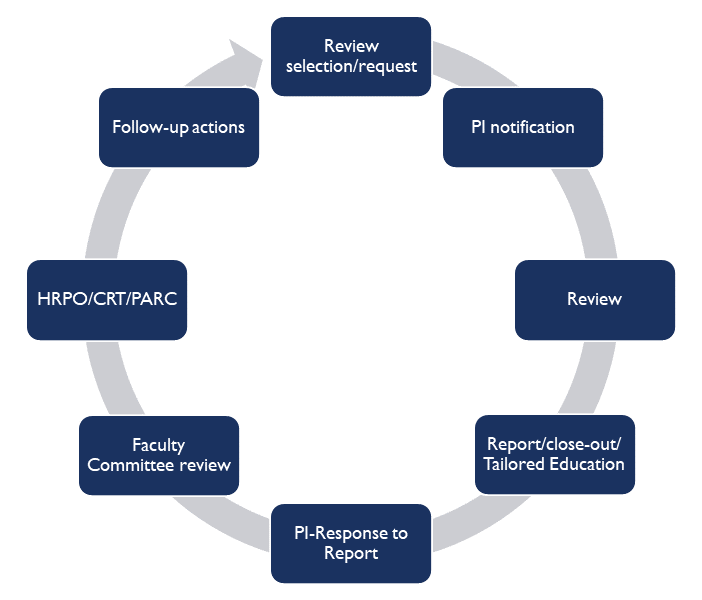
The HRQA staff will first meet with the PI and designated research staff to review the findings from the visit and provide customized education and guidance based on the review findings. A copy of the HRQA report is then sent electronically to the PI and designated research staff for review and response.
The HRQA Faculty Committee will review the report and the PI’s response to the findings, and assess whether compliance standards have been met. A copy of the report, PI response, and the HRQA Committee’s Assessment will be shared with the PI and designated research team members, the department and/or division head, the Human Research Protection Office (HRPO), the Office of the Vice Chancellor for Research (OVCR), and other compliance offices as appropriate.
Please note: Serious concerns may need to be reported directly to OVCR, HRPO, and/or other compliance offices prior to HRQA Faculty Committee Review.
Key Elements for Monitoring Adherence
There 8 key elements to consider when maintaining quality research:
I. Study Enrollment Status
- Is there adequate tracking and reporting of research participant involvement in a research study?
- Has enrollment information and participant status been reported appropriately to HRPO?
- Has tracking of participant withdrawal or screen-fail been documented appropriately?
- Are screening, enrollment, and master logs being maintained accurately and in line with your approved application & protocol?
II. Execution of Informed Consent/Assent
- Has informed consent/assent been obtained/documented according to the process approved in the myIRB application?
- Did the informed consent/assent document contain errors, including but not limited to:
- Outdated or expired documents
- Invalid or missing documents
- Answerable items left blank
- Study procedures executed prior to or without consent
- Lack of full execution of signature boxes
- Signature date discrepancies
- Consent not obtained by appropriate Legally Authorized Representative (LAR) or Legal Parent/Guardian
- Consent obtained by non-HRPO-approved person
III. Maintaining Privacy/Confidentiality
- Has the participants’ private information and study data been used, maintained, and/ or disclosed appropriately?
- Are study records being retained according to University Record Retention Policies (minimum of 6 years post study closure in myIRB)?
- Have there been any HIPAA breaches?
- Is study data being maintained according to protocol?
IV. Participant Eligibility
- Have there been any deficiencies identified with the tracking, documentation, and/or enrollment of research participants? This includes but is not limited to:
- Lack or inconsistent documentation of inclusion/exclusion criteria,
- Enrollment of ineligible participants
- Lack of documentation of the source inclusion/exclusion were verified from (per the participant, per EMR on XX/XX/XXXX value X, per interview/survey, etc.)
- Duplicate enrollment without HRPO Approval
- Enrollment of a non-HRPO approved population
V. Documentation of Interventions/Observations & Study Procedures
- Have there been any deficiencies identified with how the investigational plan was implemented and/or documented in the research record? This includes, but is not limited to:
- Deviations from the approved protocol/study plan
- Errors in study documentation (incomplete CRFs, lack of good documentation practices, etc.)
- Inconsistent or lack of documentation of study procedures
- Lack of documentation of research team member/PI involvement and oversight
VI. Safety Monitoring/Adverse Events
- Have there been deficiencies identified with the monitoring, tracking, and reporting of participant safety? This includes, but is not limited to:
- Safety monitoring not conducted or documented according to protocol
- Errors in the monitoring and/or collection of adverse and serious adverse events
- Lack of adherence to safety reporting requirements
- Lack of adherence to HRPO safety reporting requirements
VII. Regulatory Documentation/Multisite Study Management
- Have there been deficiencies identified with the maintenance of essential regulatory documentation at the local site or multisite level? This includes, but is not limited to:
- Failure to maintain essential regulatory documentation
- Discrepancies within essential regulatory documentation
- Participant payment discrepancies
VIII. Required Research Education/HIPAA Training
- Have all research team members completed the required research education (CITI Human Subjects Training, Good Clinical Practice Training, and HIPAA 101) as applicable?
Internally monitoring and self-auditing human research studies is a valuable way for researchers to ensure that participants’ rights, safety and welfare are protected and that the research is conducted in accordance with the approved research plan and documented according to best practices. Find more information and tools in the Self-Audit Toolkit.